![[sigma]](../greek/SIGMA.GIF) -adduct (sigma adduct); -bond (sigma bond); -constant (sigma constant); -orbital (sigma orbital)
-adduct (sigma adduct); -bond (sigma bond); -constant (sigma constant); -orbital (sigma orbital)
Contents
stable; stationary state; steady state (or stationary state); stepwise reaction; stereoelectronic; stereoelectronic control; stereoselectivity, stereoselective; stereospecificity, stereospecific; steric-approach control; steric effect; steric isotope effect; steric hindrance; stoichiometric number; stopped flow; strain; subjacent orbital; substituent; substituent electronegativity; substitution reaction; substrate; successor complex; suicide inhibition; superacid; superbase; suprafacial; supramolecule; Swain-Lupton equation; Swain-Scott equation; symbiosis; symproportionation; syn; synartetic acceleration; synchronization (principle of nonperfect synchronization); synchronous; -adduct (sigma adduct); -bond (sigma bond); -constant (sigma constant); -orbital (sigma orbital)
As applied to chemical species, the term expresses a thermodynamic property, which is quantitatively measured by relative molar standard Gibbs energies. A chemical species A is more stable than its isomer B if ![[Delta]](../greek/DDELTA.GIF) rGo > 0 for the (real or hypothetical) reaction A
rGo > 0 for the (real or hypothetical) reaction A ![]() B, under standard conditions. If for the two reactions
B, under standard conditions. If for the two reactions
rG1o)
Q ![]() X + Z (rG2o)
X + Z (rG2o)
rG1o > rG2o, P is more stable relative to the product Y than is Q relative to Z. Both in qualitative and quantitative usage the term stable is therefore always used in reference to some explicitly stated or implicitly assumed standard.
The term should not be used as a synonym for unreactive or "less reactive" since this confuses thermodynamics and kinetics. A relatively more stable chemical species may be more reactive than some reference species towards a given reaction partner.
(1) In quantum mechanics: A state that does not evolve with time.
(2) In kinetics: See steady state.
steady state (or stationary state)
(1) In a kinetic analysis of a complex reaction involving unstable intermediates in low concentration, the rate of change of each such intermediate is set equal to zero, so that the rate equation can be expressed as a function of the concentrations of chemical species present in macroscopic amounts. For example, assume that X is an unstable intermediate in the reaction sequence:
![[reversible arrow]](POgif/RATE6.GIF) X
X
X + C ![[arrow]](POgif/RATE7.GIF) D
D
Conservation of mass requires that:
which, since [A]0 is constant, implies:
Since [X] is negligibly small, the rate of formation of D is essentially equal to the rate of disappearance of A, and the rate of change of [X] can be set equal to zero. Applying the steady state approximation (d[X]/dt = 0) allows the elimination of [X] from the kinetic equations, whereupon the rate of reaction is expressed:
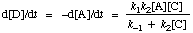
Note: The steady-state approximation does not imply that [X] is even approximately constant, only that its absolute rate of change is very much smaller than that of [A] and [D]. Since according to the reaction scheme d[D]/dt = k2[X][C], the assumption that [X] is constant would lead, for the case in which C is in large excess, to the absurd conclusion that formation of the product D will continue at a constant rate even after the reactant A has been consumed.
(2) In a stirred flow reactor a steady state implies a regime so that all concentrations are independent of time.
A chemical reaction with at least one reaction intermediate and involving at least two consecutive elementary reactions.
See also composite reaction.
Pertaining to the dependence of the properties (especially the energy) of a molecular entity in a particular electronic state (or of a transition state) on relative nuclear geometry. The electronic ground state is usually considered, but the term can apply to excited states as well. Stereoelectronic effects are ascribed to the different alignment of electronic orbitals in different arrangements of nuclear geometry.
Control of the nature of the products of a chemical reaction (or of its rate) by stereoelectronic factors. The term is usually applied in the framework of an orbital approximation. The variations of molecular orbital energies with relative nuclear geometry (along a reaction coordinate) are then seen as consequences of variations in basis-orbital overlaps.
stereoselectivity, stereoselective
Stereoselectivity is the preferential formation in a chemical reaction of one stereoisomer over another. When the stereoisomers are enantiomers, the phenomenon is called enantioselectivity and is quantitatively expressed by the enantiomer excess; when they are diastereoisomers, it is called diastereoselectivity and is quantitatively expressed by the diastereomer excess. Reactions are termed (100%) stereoselective if the discrimination is complete or partially (x%) stereoselective if one product predominates. The discrimination may also be referred to semiquantitatively as high or low stereoselectivity. ELIEL (1962); IUPAC STEREOCHEMICAL TERMINOLOGY (1993).
stereospecificity, stereospecific
(1) A reaction is termed stereospecific if starting materials differing only in their configuration are converted into stereoisomeric products. According to this definition, a stereospecific process is necessarily stereoselective but not all stereoselective processes are stereospecific. Stereospecificity may be total (100%) or partial. The term is also applied to situations where reaction can be performed with only one stereoisomer. For example the exclusive formation of trans-1,2-dibromocyclohexane upon bromination of cyclohexene is a stereospecific process, although the analogous reaction with (E)-cyclohexene has not been performed.
(2) The term has also been applied to describe a reaction of very high stereoselectivity, but this usage is unnecessary and is discouraged.
ELIEL (1962). [For the use of the term "stereospecific polymerization" see IUPAC POLYMERS (1981)]
Control of stereoselectivity of a reaction by steric hindrance towards attack of the reagent, which is directed to the less hindered face of the molecule. Partial bond making is strong enough at the transition state for steric control to take place. This suggests that the transition state should not be close to products. See also product development control.
The effect on a chemical or physical property (structure, rate or equilibrium constant) upon introduction of substituents having different steric requirements. The steric effect in a reaction is ascribed to the difference in steric energy between, on the one hand, reactants and, on the other hand, a transition state, (or products). A steric effect on a rate process may result in a rate increase ("steric acceleration") or a decrease ("steric retardation"). (The adjective "steric" is not to be confused with stereochemical.)
Steric effects arise from contributions ascribed to strain as the sum of (1) non-bonded repulsions, (2) bond angle strain, and (3) bond stretches or compressions.
For the purpose of correlation analysis or linear free-energy relations various scales of steric parameters have been proposed, notably A values, Taft's Es and Charton's ![[upsilon]](../greek/UPSILON.GIF) scales.
scales.
In a reactant molecule RY and an appropriate reference molecule R0Y, the "primary steric effect" of R is the direct result of differences in compressions which occur because R differs from R0 in the vicinity of the reaction centre Y. A "secondary steric effect" involves the differential moderation of electron delocalization by non-bonded compressions.
Some authors make a distinction between "steric" effects attributed to van der Waals repulsions alone, and "strain" effects, attributed to deviations of bond angles from "ideal" values. CHARTON (1987). See Taft equation, van der Waals forces.
The original term for a steric effect arising from crowding of substituents.
See rate of reaction.
A technique for following the kinetics of reactions in solution (usually in the millisecond time range) in which two reactant solutions are rapidly mixed by being forced through a mixing chamber. The flow of the mixed solution along a uniform tube is then suddenly arrested. At a fixed position along the tube the solution is monitored (as a function of time following the stoppage of the flow) by some method with a rapid response (e.g. photometry). See mixing control.
Strain is present in a molecular entity or transition structure if the energy is enhanced because of unfavourable bond lengths, bond angles, or dihedral angles ("torsional strain") relative to a standard.
It is quantitatively defined as the standard enthalpy of a structure relative to a strainless structure (real or hypothetical) made up from the same atoms with the same types of bonding. (The enthalpy of formation of cyclopropane is 53.6 kJ mol-1, whereas the enthalpy of formation based on three "normal" methylene groups, from acyclic models, is -62 kJ mol-1. On this basis cyclopropane is destabilized by ca. 115 kJ mol-1 of strain energy.) See molecular mechanics calculation.
The Next-to-Highest Occupied Molecular Orbital ("NHOMO", also called "HOMO-") and the Second Lowest Unoccupied Molecular Orbital (SLUMO). Subjacent orbitals are sometimes found to play an important role in the interpretation of molecular interactions according to the frontier orbital approach. BERSON (1972).
An atom or group of bonded atoms that can be considered to have replaced a hydrogen atom (or two hydrogen atoms in the special case of bivalent groups) in a parent molecular entity (real or hypothetical).
See electronegativity.
A reaction, elementary or stepwise, in which one atom or group in a molecular entity is replaced by another atom or group. For example,
A chemical species, the reaction of which with some other chemical reagent is under observation (e.g., a compound that is transformed under the influence of a catalyst). The term should be used with care. Either the context or a specific statement should always make it clear which chemical species in a reaction is regarded as the substrate. See also transformation.
The radical ion pair which forms by the transfer of an electron from the donor D to the acceptor A after these species have diffused together to form the precursor or encounter complex:
See mechanism-based inhibition.
A medium having a high acidity, generally greater than that of 100 wt.-% sulfuric acid. The common superacids are made by dissolving a powerful Lewis acid (e.g. SbF5) in a suitable Brønsted acid, such as HF or HSO3F. (An equimolar mixture of HSO3F and SbF5 is known by the trade name "magic acid".)
In a biochemical context "superacid catalysis" is sometimes used to denote catalysis by metal ions analogous to catalysis by hydrogen ions.
By analogy, a compound having a very high basicity, such as lithium diisopropylamide, is called a "superbase". GILLESPIE (1968); OLAH (1985); OLAH and OLAH (1970).
See superacid.
See antarafacial.
A system of two or more molecular entities held together and organized by means of intermolecular (noncovalent) binding interactions. LEHN (1993).
A dual parameter approach to the correlation analysis of substituent effects, which involves a field constant (F) and a resonance constant (R). The original treatment was modified later. SWAIN and LUPTON (1968); SWAIN, UNGER, ROSENQUIST and SWAIN (1983).
The procedure has been considerably applied, but also much criticized (see REYNOLDS and TOPSOM (1984); HOEFNAGEL, OOSTERBEEK and WEPSTER (1984); CHARTON (1984); SWAIN (1984); HANSCH, LEO, and TAFT (1991)).
The linear free-energy relation of the form
applied to the variation of reactivity of a given electrophilic substrate towards a series of nucleophilic reagents. n is characteristic of the reagent (i.e. a measure of its nucleophilicity) and s is characteristic of the substrate (i.e. a measure of its sensitivity to the nucleophilicity of the reagent). A scale of n values is based on the rate coefficients k for the reaction of methyl bromide with nucleophiles in water at 25 oC, s being defined as 1.00 for these reactions and n being defined as 0.00 for the hydrolysis of methyl bromide. (Other scales have been devised.) SWAIN and SCOTT (1953).
The term was originally applied to describe the maximum flocking of either hard or soft ligands in the same complexes. For hydrocarbon molecules, symbiosis implies that those containing a maximum number of C-H bonds (e.g. CH4) or C-C bonds (e.g. Me4C) are the most stable. HO (1977).
Synonymous with comproportionation.
See anti.
See neighbouring group participation.
synchronization (principle of nonperfect synchronization)
This principle applies to reactions in which there is a lack of synchronization between bond formation or bond rupture and other primitive changes that affect the stability of products and reactants, such as resonance, solvation, electrostatic, hydrogen bonding and polarizability effects. The principle states that a product-stabilizing factor whose development lags behind bond changes at the transition state, or a reactant-stabilizing factor whose loss is ahead of bond changes at the transition state, increases the intrinsic barrier and decreases the "intrinsic rate constant" of a reaction. For a product-stabilizing factor whose development is ahead of bond changes, or reactant factors whose loss lags behind bond changes, the opposite relations hold. The reverse effects are observable for factors that destabilize a reactant or product. BERNASCONI (1992). See also imbalance, synchronous.
A concerted process in which the primitive changes concerned (generally bond rupture and bond formation) have progressed to the same extent at the transition state is said to be synchronous. The term figuratively implies a more or less synchronized progress of the changes. However, the progress of the bonding change (or other primitive change) has not been defined quantitatively in terms of a single parameter applicable to different bonds or different bonding changes. The concept is therefore in general only qualitatively descriptive and does not admit an exact definition except in the case of concerted processes involving changes in two identical bonds. See also imbalance.
The product formed by the attachment of an electrophilic or nucleophilic entering group or of a radical to a ring carbon of an aromatic species so that a new sigma bond is formed and the original conjugation is disrupted. (This has generally been called a "-complex", but adduct is more appropriate than complex according to the definitions given.) The term may also be used for analogous adducts to unsaturated (and conjugated) systems in general. See also Meisenheimer adduct.
See sigma, pi.
Specifically the substituent constant for meta- and for para-substituents in benzene derivatives as defined by Hammett on the basis of the ionization constant of a substituted benzoic acid in water at 25 oC, i.e. lg(Ka/Kao), where Ka is the ionization constant of a m- or p-substituted benzoic acid and Kao that of benzoic acid itself.
The term is also used as a collective description for related electronic substituent constants based on other standard reaction series, of which, +, - and o are typical; also constants which represent dissected electronic effects such as I and R. For this purpose it might be better always to spell out the term in full, i.e. as "Hammett sigma constant", and restrict -constants to the scale of substituent constants which is based on benzoic acid.
A large positive -value implies high electron-withdrawing power by inductive and/or resonance effect, relative to H; a large negative -value implies high electron-releasing power relative to H. CHAPMAN and SHORTER (1972, 1978); JOHNSON (1973); SHORTER (1973). See also Hammett equation, ![[rho]](../greek/RHO.GIF) -value, Taft equation.
-value, Taft equation.
See sigma, pi.
BERSON, J. A. (1972), Acc. Chem. Res., 5, 406-414.
CHARTON, M. (1984), J. Org. Chem., 49, 1997-2001.
CHARTON, M. (1987), Progr. Phys. Org. Chem., 16, 287-315.
ELIEL, E. L. (1962), "Stereochemistry of Carbon Compounds", McGraw Hill, New York.
GILLESPIE, R. J. (1968), Acc. Chem. Res., 1, 202-209.
HANSCH, C., LEO, A., and TAFT, R. W. (1991), Chem. Rev., 91, 165-195.
HOEFNAGEL, A. J., OOSTERBEEK, W., and WEPSTER, B. M. (1984), J. Org. Chem. , 49, 1993-1997.
*IUPAC STEREOCHEMICAL TERMINOLOGY (1993). IUPAC: Organic Chemistry Division: Basic Terminology of Stereochemistry. IDCNS and public review. Now published as Basic Terminology of Stereochemistry (IUPAC Recommendations 1996) in Pure Appl. Chem., 68, 2193-2222 (1996).
JOHNSON, C. D. (1973), "The Hammett Equation", Cambridge University Press.
LEHN, J.-M. (1993), Science, 260, 1762-1763.
OLAH, G. A. (1985), "Superacids". Wiley, New York.
REYNOLDS, W. F., and TOPSOM, R. D. (1984), J. Org. Chem., 49, 1989-1992.
SWAIN, C. G. (1984), J. Org. Chem., 49, 2005-2010.
SWAIN. C. G., and LUPTON, E. C. (1968), J. Am. Chem. Soc., 90, 4328-4337.
SWAIN, C. G., and SCOTT, C. B. (1953), J. Am. Chem. Soc., 75, 141-147.
Return to home page for Glossary of terms used in Physical Organic Chemistry.