![[mu]](../greek/MU.GIF) (mu) and
(mu) and ![[italic mu]](../greek/MUITAL.GIF)
Contents
macroscopic diffusion control; magic acid; magnetic equivalence; magnetization transfer; Marcus equation; Markownikoff rule; mass-law effect; matrix isolation; mean lifetime; mechanism; mechanism-based inhibition; medium; Meisenheimer adduct; melting point (corrected/uncorrected); mesolytic cleavage; mesomeric effect; mesomerism; mesophase; metastable (chemical species); metathesis; + methylene; + methylidyne; micellar catalysis; micelle; Michaelis-Menten kinetics; microscopic chemical event; microscopic diffusion control (encounter control); microscopic reversibility, principle of; migration; migratory aptitude; migratory insertion; minimum structural change, principle of; mixing control; Möbius aromaticity; moiety; molecular entity; molecularity; molecular mechanics calculation; molecular metal; molecular orbital; molecular rearrangement; molecule; More O'Ferrall-Jencks diagram; multi-centre bond; + multi-centre reaction; multident; (mu) and
See mixing control.
See superacid.
Nuclei having the same resonance frequency in nuclear magnetic resonance spectroscopy and also identical spin-spin interactions with the nuclei of a neighbouring group are magnetically equivalent. The spin-spin interaction between magnetically equivalent nuclei does not appear, and thus has no effect on the multiplicity of the respective NMR signals. Magnetically equivalent nuclei are necessarily also chemically equivalent, but the reverse is not necessarily true.
NMR method for determining kinetics of chemical exchange by perturbing the magnetization of nuclei in a particular site or sites and following the rate at which magnetic equilibrium is restored. The most common perturbations are saturation and inversion, and the corresponding techniques are often called "saturation transfer" and "selective inversion-recovery". See also saturation transfer.
A general expression which correlates the Gibbs energy of activation (![[Delta]](../greek/DDELTA.GIF)
![[double dagger]](../greek/DDAGGER.GIF) G) with the driving force rGo')
G) with the driving force rGo')
G = (![[lambda]](../greek/LAMBDA.GIF) /4)(1 + rGo'/)2
/4)(1 + rGo'/)2
where is the reorganization energy and rGo' is the standard free energy of the reaction corrected for the electrostatic work required to bring the reactants together. /4 is the intrinsic barrier of the reaction. Originally developed for outer-sphere electron transfer reactions, the Marcus equation has later been applied also to atom and group transfer reactions. MARCUS (1964); ALBERY (1980).
"In the addition of hydrogen halides to unsymmetrically constituted [unsaturated] hydrocarbons, the halogen atom becomes attached to the carbon bearing the lesser number of hydrogen atoms." Originally formulated by Markownikoff (Markovnikov) to generalize the orientation in additions of hydrogen halides to simple alkenes, this rule has been extended to polar addition reactions as follows. " In the heterolytic addition of a polar molecule to an alkene or alkyne, the more electronegative (nucleophilic) atom (or part) of the polar molecule becomes attached to the carbon atom bearing the smaller number of hydrogen atoms."
This is an indirect statement of the common mechanistic observation, that the more electropositive (electrophilic) atom (or part) of the polar molecule becomes attached to the end of the multiple bond that would result in the more stable carbenium ion (whether or not a carbenium ion is actually formed as a reaction intermediate in the addition reaction). Addition in the opposite sense is commonly called "anti-Markovnikov addition". MARKOWNIKOFF (1870).
At equilibrium, the product of the activities (or concentrations) of the reacting species is constant. Thus for the equilibrium
![[alpha]](../greek/ALPHA.GIF) A +
A + ![[beta]](../greek/BETA.GIF) B
B ![[reversible arrow]](../greek/REVARR.GIF)
![[gamma]](../greek/GAMMA.GIF) C +
C + ![[delta]](../greek/DELTA.GIF) D
D
K = [C][D]/[A][B]
GULDBERG and WAAGE (1879). See also common-ion effect, equilibrium.
A term which refers to the isolation of a reactive or unstable species by dilution in an inert matrix (argon, nitrogen, etc.), usually condensed on a window or in an optical cell at low temperature, to preserve its structure for identification by spectroscopic or other means. IUPAC ATMOSPHERIC GLOSSARY (1990).
See lifetime.
A detailed description of the process leading from the reactants to the products of a reaction, including a characterization as complete as possible of the composition, structure, energy and other properties of reaction intermediates, products, and transition states. An acceptable mechanism of a specified reaction (and there may be a number of such alternative mechanisms not excluded by the evidence) must be consistent with the reaction stoichiometry, the rate law, and with all other available experimental data, such as the stereochemical course of the reaction. Inferences concerning the electronic motions which dynamically interconvert successive species along the reaction path (as represented by curved arrows, for example) are often included in the description of a mechanism.
It should be noted that for many reactions all this information is not available and the suggested mechanism is based on incomplete experimental data. It is not appropriate to use the term mechanism to describe a statement of the probable sequence in a set of stepwise reactions. That should be referred to as a reaction sequence, and not a mechanism. See also Gibbs energy diagram.
Irreversible inhibition of an enzyme due to its catalysis of the reaction of an artificial substrate. Also called "suicide inhibition".
The phase (and composition of the phase) in which chemical species and their reactions are studied in a particular investigation.
A cyclohexadienyl derivative formed as Lewis adduct from a nucleophile (Lewis base) and an aromatic or heteroaromatic compound, also called Jackson-Meisenheimer adduct. In earlier usage the term "Meisenheimer complex" was restricted to the typical Meisenheimer alkoxide adducts of nitro-substituted aromatic ethers, e.g.,
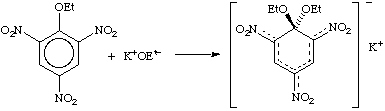
Analogous cationic adducts, such as
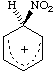
considered to be reaction intermediates in electrophilic aromatic substitution reactions, are called "Wheland intermediates", and sometimes, inappropriately, ![[sigma]](../greek/SIGMA.GIF) -complexes. JACKSON and GAZZOLO (1900); BUNCEL, CRAMPTON, STRAUSS and TERRIER. (1984). See also -adduct.
-complexes. JACKSON and GAZZOLO (1900); BUNCEL, CRAMPTON, STRAUSS and TERRIER. (1984). See also -adduct.
melting point (corrected/uncorrected)
The term originally signified that a correction was made (not made) for the emergent stem of the thermometer. In current usage it often means that the accuracy of the thermometer was (was not) verified. This current usage is inappropriate and should be abandoned.
Cleavage of a bond in a radical ion whereby a radical and an ion are formed. The term reflects the mechanistic duality of the process, which can be viewed as homolytic or heterolytic depending on how the electrons are attributed to the fragments. See MASLAK and NARVAEZ (1990).
The effect (on reaction rates, ionization equilibria, etc.) attributed to a substituent due to overlap of its p or pi orbitals with the p or pi orbitals of the rest of the molecular entity. Delocalization is thereby introduced or extended, and electronic charge may flow to or from the substituent. The effect is symbolized by M.
Strictly understood, the mesomeric effect operates in the ground electronic state of the molecule. When the molecule undergoes electronic excitation or its energy is increased on the way to the transition state of a chemical reaction, the mesomeric effect may be enhanced by the electromeric effect, but this term is not much used, and the mesomeric and electromeric effects tend to be subsumed in the term resonance effect of a substituent. See also electronic effect, field effect, inductive effect.
Essentially synonymous with resonance. The term is particularly associated with the picture of pi electrons as less localized in an actual molecule than in a Lewis formula.
The term is intended to imply that the correct representation of a structure is intermediate between two or more Lewis formulae. See also aromatic (2), delocalization.
The phase of a liquid crystalline compound between the crystalline and the isotropic liquid phase.
See transient (chemical species).
A bimolecular process formally involving the exchange of a bond (or bonds) between similar interacting chemical species so that the bonding affiliations in the products are identical (or closely similar) to those in the reactants. For example:
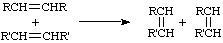
(The term has its origin in inorganic chemistry with a different meaning, but this older usage is not applicable in physical organic chemistry.)
See carbene.
See carbyne.
The acceleration of a chemical reaction in solution by the addition of a surfactant at a concentration higher than its critical micelle concentration so that the reaction can proceed in the environment of surfactant aggregates (micelles). (Rate enhancements may be due, for example, to higher concentration of the reactants in that environment, more favourable orientation and solvation of the species, or enhanced rate constants in the micellar pseudophase of the surfactant aggregate.) Micelle formation can also lead to a decreased reaction rate. See also catalyst.
Surfactants in solution are often association colloids, that is, they tend to form aggregates of colloidal dimensions, which exist in equilibrium with the molecules or ions from which they are formed. Such aggregates are termed micelles. See also inverted micelle. IUPAC MANUAL APPENDIX II (1972).
The dependence of an initial rate of reaction upon the concentration of a substrate S that is present in large excess over the concentration of an enzyme or other catalyst (or reagent) E with the appearance of saturation behaviour following the Michaelis-Menten equation,
where v is the observed initial rate, V is its limiting value at substrate saturation (i.e., [S] >> Km), and Km the substrate concentration when v = V/2. The definition is experimental, i.e., it applies to any reaction that follows an equation of this general form. The symbols Vma or vma are sometimes used for V.
The parameters V and Km (the "Michaelis constant") of the equation can be evaluated from the slope and intercept of a linear plot of v-1 against [S]-1 (a "Lineweaver-Burk plot") or from slope and intercept of a linear plot of v against h/[S] ("Eadie-Hofstee plot").
A Michaelis-Menten equation is also applicable to the condition where E is present in large excess, in which case the concentration [E] appears in the equation instead of [S].
The term has sometimes been used to describe reactions that proceed according to the scheme
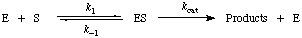
in which case Km = (k-1 + kcat)/k1 (Briggs-Haldane conditions). It has more usually been applied only to the special case in which k-1 >> kcat and Km = k-1/k1 = Ks; in this case Km is a true dissociation constant (Michaelis-Menten conditions). See also rate-determining step.
See chemical reaction, molecularity.
microscopic diffusion control (encounter control)
The observable consequence of the limitation that the rate of a bimolecular chemical reaction in a homogeneous medium cannot exceed the rate of encounter of the reacting molecular entities.
If (hypothetically) a bimolecular reaction in a homogeneous medium occurred instantaneously when two reactant molecular entities made an encounter, the rate of reaction would be an encounter-controlled rate, determined solely by rates of diffusion of reactants. Such a hypothetical "fully diffusion controlled rate" is also said to correspond to "total microscopic diffusion control", and represents the asymptotic limit of the rate of reaction as the rate constant for the chemical conversion of the encounter pair into product (or products) becomes large relative to the rate constant for separation (or dissociation) of the encounter pair.
"Partial microscopic diffusion control" is said to operate in a homogeneous reaction when the rates of chemical conversion and of separation are comparable. (The degree of microscopic diffusion control cannot usually be determined with any precision.) See also mixing control.
microscopic reversibility, principle of
In a reversible reaction, the mechanism in one direction is exactly the reverse of the mechanism in the other direction. This does not apply to reactions that begin with a photochemical excitation. See also chemical reaction, detailed balancing.
(1) The (usually intramolecular) transfer of an atom or group during the course of a molecular rearrangement.
(2) The movement of a bond to a new position, within the same molecular entity, is known as "bond migration".
Allylic rearrangements, e.g.,
exemplify both types of migration.
The term is applied to characterize the relative tendency of a group to participate in a rearrangement. In nucleophilic rearrangements (migration to an electron-deficient centre), the migratory aptitude of a group is loosely related to its capacity to stabilize a partial positive charge, but exceptions are known, and the position of hydrogen in the series is often unpredictable.
A combination of migration and insertion. The term is mainly used in organometallic chemistry.
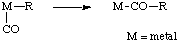
minimum structural change, principle of
The experimental limitation of the rate of reaction in solution by the rate of mixing of solutions of the two reactants. It can occur even when the reaction rate constant is several powers of 10 less than that for an encounter-controlled rate. Analogous (and even more important) effects of the limitation of reaction rates by the speed of mixing are encountered in heterogeneous (solid/liquid, solid/gas, liquid/gas) systems. See also microscopic diffusion control, stopped flow.
A monocyclic array of orbitals in which there is a single out-of-phase overlap (or, more generally, an odd number of out-of-phase overlaps) reveals the opposite pattern of aromatic character to Hückel systems; with 4n electrons it is stabilized (aromatic), whereas with 4n + 2 it is destabilized (antiaromatic). In the excited state 4n + 2 Möbius pi-electron systems are stabilized, and 4n systems are destabilized. No examples of ground-state Möbius pi systems are known, but the concept has been applied to transition states of pericyclic reactions [see aromatic (3)].
The name is derived from the topological analogy of such an arrangement of orbitals to a Möbius strip. HEILBRONNER (1964); ZIMMERMAN (1971). See also Hückel (4n + 2) rule.
In physical organic chemistry moiety is generally used to signify part of a molecule, e.g. in an ester R1COOR2 the alcohol moiety is R2O. The term should not be used for a small fragment of a molecule.
Any constitutionally or isotopically distinct atom, molecule, ion, ion pair, radical, radical ion, complex, conformer etc., identifiable as a separately distinguishable entity.
Molecular entity is used in this glossary as a general term for singular entities, irrespective of their nature, while chemical species stands for sets or ensembles of molecular entities. Note that the name of a compound may refer to the respective molecular entity or to the chemical species, e.g. methane, may mean a single molecule of CH4 (molecular entity) or a molar amount, specified or not (chemical species), participating in a reaction.
The degree of precision necessary to describe a molecular entity depends on the context. For example "hydrogen molecule" is an adequate definition of a certain molecular entity for some purposes, whereas for others it is necessary to distinguish the electronic state and/or vibrational state and/or nuclear spin, etc. of the hydrogen molecule.
The number of reactant molecular entities that are involved in the "microscopic chemical event" constituting an elementary reaction. (For reactions in solution this number is always taken to exclude molecular entities that form part of the medium and which are involved solely by virtue of their solvation of solutes.) A reaction with a molecularity of one is called "unimolecular", one with a molecularity of two "bimolecular" and of three "termolecular". See also chemical reaction, order of reaction.
molecular mechanics calculation
An empirical calculational method intended to give estimates of structures and energies for conformations of molecules. The method is based on the assumption of "natural" bond lengths and angles, deviation from which leads to strain, and the existence of torsional interactions and attractive and/or repulsive van der Waals and dipolar forces between non-bonded atoms. The method is also called "(empirical) force-field calculations". BURKERT and ALLINGER (1982).
A non-metallic material whose properties resemble those of metals, usually following oxidative doping; e.g. polyacetylene following oxidative doping with iodine.
A one-electron wavefunction describing an electron moving in the effective field provided by the nuclei and all other electrons of a molecular entity of more than one atom. Such molecular orbitals can be transformed in prescribed ways into component functions to give "localized molecular orbitals". Molecular orbitals can also be described, in terms of the number of nuclei (or "centres") encompassed, as two-centre, multi-centre, etc. molecular orbitals, and are often expressed as a linear combination of atomic orbitals.
An orbital is usually depicted by sketching contours on which the wavefunction has a constant value (contour map) or by indicating schematically the envelope of the region of space in which there is an arbitrarily fixed high (say 96%) probability of finding the electron occupying the orbital, giving also the algebraic sign (+ or -) of the wavefunction in each part of that region.
The term is traditionally applied to any reaction that involves a change of connectivity (sometimes including hydrogen), and violates the so-called "principle of minimum structural change". According to this oversimplified principle, chemical species do not isomerize in the course of a transformation, e.g. substitution, or the change of a functional group of a chemical species into a different functional group is not expected to involve the making or breaking of more than the minimum number of bonds required to effect that transformation. For example, any new substituents are expected to enter the precise positions previously occupied by displaced groups.
The simplest type of rearrangement is an intramolecular reaction in which the product is isomeric with the reactant (one type of "intramolecular isomerization"). An example is the first step of the Claisen rearrangement:

The definition of molecular rearrangement includes changes in which there is a migration of an atom or bond (unexpected on the basis of the principle of minimum structural change), as in the reaction
where the rearrangement stage can formally be represented as the "1,2-shift" of hydride between adjacent carbon atoms in the carbocation
Such migrations occur also in radicals, e.g.:
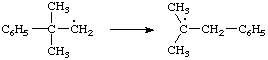
The definition also includes reactions in which an entering group takes up a different position from the leaving group, with accompanying bond migration. An example of the latter type is the "allylic rearrangement":
A distinction is made between "intramolecular rearrangements" (or "true molecular rearrangements") and "intermolecular rearrangements" (or "apparent rearrangements"). In the former case the atoms and groups that are common to a reactant and a product never separate into independent fragments during the rearrangement stage (i.e. the change is intramolecular), whereas in an "intermolecular rearrangement" a migrating group is completely free from the parent molecule and is re-attached to a different position in a subsequent step, as in the Orton reaction:
An electrically neutral entity consisting of more than one atom (n > 1). Rigorously, a molecule, in which n > 1 must correspond to a depression on the potential energy surface that is deep enough to confine at least one vibrational state. See also molecular entity.
Visualization of the potential energy surfaces for a reacting system, as a function of two chosen coordinates. It is particularly useful to discuss structural effects on the transition state geometry for processes occurring either by stepwise or concerted routes. The use of such diagrams, first suggested for elimination reactions (MORE O'FERRALL (1970)), was later extended to acid-base catalysis and to certain other reactions (JENCKS (1972, 1980)).
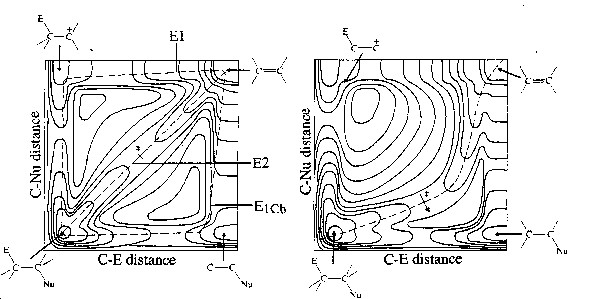
Figure. More O'Ferrall-Jencks diagrams of energy contours for -elimination reactions as a function of lengths of the two bonds broken. Reaction coordinates follow the dotted lines. Left: E2 mechanism with "central" character, simultaneous fission of C-Nu and C-H bonds. Right: The result of stabilization of the carbanion, Nu-C-C-; a continuation of this trend would result in a switch to the E1cb mechanism (Reproduced from N. S. ISAACS, "Physical Organic Chemistry", Longman Scientific, Essex, UK, (1987), with permission of Longman Scientific).
Structural changes influencing vibrational modes of the transition states cause changes in transition state geometry. Changes in the direction of the reaction coordinate (reactant or product stabilizing- or destabilizing factors) cause changes according to the Hammond principle. Structural changes perpendicular to the reaction coordinate (anti-Hammond effects, perpendicular effects) cause changes opposite to the Hammond behaviour, i.e., the easier the process related to the structural change, the more advanced it will be at the transition state. See WINEY and THORNTON (1975).
Representation of some molecular entities solely by localized two-electron two-centre bonds appears to be unsatisfactory. Instead, multi-centre bonds have to be considered in which electron pairs occupy orbitals encompassing three or more atomic centres. Examples include the three-centre bonds in diborane, the delocalized pi bonding of benzene, and bridged carbocations.
A synonym for pericyclic reaction. The number of "centres" is the number of atoms not bonded initially, between which single bonds are breaking or new bonds are formed in the transition state. This number does not necessarily correspond to the ring size of the transition state for the pericyclic reaction. Thus, a Diels-Alder reaction is a "four-centre reaction". This terminology has largely been superseded by the more detailed one developed for the various pericyclic reactions. See cycloaddition, sigmatropic rearrangement.
See ambident.
Notation for a ligand (prefix) that bridges two or more metal centres. The symbol is used for dipole moments.
ALBERY, W. J. (1980), Annu. Rev. Phys. Chem., 31, 227-263.
GULDBERG, C. M., and WAAGE, P. (1879), J. prakt. Chem., 19, 69-114.
HEILBRONNER, E. (1964), Tetrahedron Lett., 1923-1928.
ISAACS, N. S. (1987), "Physical Organic Chemistry", Longman, Essex.
JACKSON, C. J., GAZZOLO, F. H. (1900),Am. Chem. J., 23, 376-396.
JENCKS, W. P. (1972), Chem. Rev., 72, 705-718.
JENCKS, W. P. (1980), Acc. Chem. Res., 13, 161-169.
MARCUS, R. A. (1964), Annu. Rev. Phys. Chem., 15, 155-196.
MARKOWNIKOFF, W. (1870), Justus Liebigs Ann. Chem., 153, 228-259 (p 256).
MASLAK, P., and NARVAEZ, J. N. (1990), Angew. Chem., Int. Ed. Engl., 29, 283-285.
MORE O'FERRALL, R. A. (1970), J. Chem. Soc. (B), 274-277.
WINEY, D. A., and THORNTON, E. R. (1975), J. Am. Chem. Soc., 97, 3102-3108.
ZIMMERMAN, H. E. (1971), Acc. Chem. Res., 4, 272-280.
Return to home page for Glossary of terms used in Physical Organic Chemistry.