Contents
fast-atom bombardment (FAB) mass spectroscopy; field effect; flash vacuum pyrolysis (FVP); fluxional; force-field calculations; fractionation factor, isotopic; fragmentation; free radical; frontier orbitals; functional group; gas-phase acidity; gas-phase basicity; geminate pair; geminate recombination; general acid catalysis; general base catalysis; Gibbs energy diagram; Gibbs energy of activation (standard free energy of activation); ground state; group; Grunwald-Winstein equation; guest
A method in which ions are produced in a mass spectrometer from nonvolatile or thermally fragile organic molecules by bombarding the compound in the condensed phase with energy-rich neutral particles. IUPAC MASS SPECTROSCOPY (1991).
An experimentally observable effect symbolized by F (on reaction rates, etc.) of intramolecular coulombic interaction between the centre of interest and a remote unipole or dipole, by direct action through space rather than through bonds. The magnitude of the field effect (or "direct effect") depends on the unipolar charge/dipole moment, orientation of dipole, shortest distance between the centre of interest and the remote unipole or dipole, and on the effective dielectric constant. An approach to its calculation was described by KIRKWOOD and WESTHEIMER (1938) and has been elaborated in more recent years. See also electronic effect, inductive effect, polar effect.
Thermal reaction of a molecule by exposing it to a short thermal shock at high temperature, usually in the gas phase.
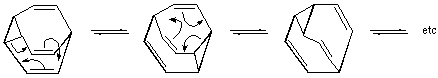
A chemical species is said to be fluxional if it undergoes rapid degenerate rearrangements (generally detectable by methods which allow the observation of the behaviour of individual nuclei in a rearranged chemical species, e.g. NMR, X-ray).
Example: Bullvalene (1 209 600 interconvertible arrangements of the ten CH groups).
The term is also used to designate positional change among ligands of complex compounds and organometallics. In these cases, the change is not necessarily degenerate. See also valence tautomerization.
See molecular mechanics calculation.
fractionation factor, isotopic
The ratio (x1/x2)A/(x1/x2)B, where x is the abundance, expressed as the atom fraction of the isotope distinguished by the subscript numeral, when the two isotopes are equilibrated between two different chemical species A and B (or between specific sites A and B in the same or different chemical species). The term is most commonly met in connection with deuterium solvent isotope effects, when the fractionation factor ![[phi]](../greek/PHI.GIF) expresses the ratio
expresses the ratio
= (xD/xH)solute/(xD/xH)solventfor the exchangeable hydrogen atoms in the chemical species (or sites) concerned. The concept is also applicable to transition states. GOLD (1969).
(1) The heterolytic cleavage of a molecule according to the general reaction
where a-b is an electrofuge and X is a nucleofuge (which may emerge from the reaction in combined form), and the middle group affords the unsaturated fragment c=d. For example,
(2) The breakdown of a radical into a diamagnetic molecule or ion and a smaller radical, e.g.,
[ArBr]. - ![]() Ar. + Br- (solution)
Ar. + Br- (solution)
(3) The breakdown of a radical ion in a mass spectrometer or in solution, forming an ion of lower molar mass and a radical, e.g.,
See radical.
The Highest-energy Occupied Molecular Orbital (HOMO) (filled or partly filled) and Lowest-energy Unoccupied Molecular Orbital (LUMO) (completely or partly vacant) of a molecular entity. Examination of the mixing of frontier molecular orbitals of reacting molecular entities affords an approach to the interpretation of reaction behaviour; this constitutes a simplified perturbation molecular orbital theory of chemical behaviour. FUKUI, YONEZAWA, and SHINGU (1952); FLEMING (1976). See also SOMO, subjacent orbital.
Organic compounds are thought of as consisting of a relatively unreactive backbone, for example a chain of sp3 hybridized carbon atoms, and one or several functional groups. The functional group is an atom, or a group of atoms that have similar chemical properties whenever it occurs in different compounds. It defines the characteristic physical and chemical properties of families of organic compounds.
The negative of the Gibbs energy (![[Delta]](../greek/DDELTA.GIF) Gro) change for the reaction
Gro) change for the reaction
in the gas phase. BARTMESS and MC IVER (1979).
The negative of the Gibbs energy (Gro) change associated with the reaction
in the gas phase. Also called absolute or intrinsic basicity. See also proton affinity.
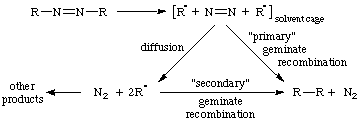
Pair of molecular entities in close proximity in solution within a solvent cage and resulting from reaction (e.g. bond scission, electron transfer, group transfer) of a precursor that constitutes a single kinetic entity. See also ion pair, radical pair.
The reaction with each other of two transient molecular entities produced from a common precursor in solution. If reaction occurs before any separation by diffusion has occurred, this is termed "primary geminate recombination". If the mutually reactive entities have been separated, and come together by diffusion, this is termed "secondary geminate recombination".
The catalysis of a chemical reaction by a series of Brønsted acids (which may include the solvated hydrogen ion) so that the rate of the catalysed part of the reaction is given by ![[Sigma]](../greek/SSIGMA.GIF) kHA[HA] multiplied by some function of substrate concentrations. (The acids HA are unchanged by the overall reaction.) General catalysis by acids can be experimentally distinguished from specific catalysis by hydrogen cations (hydrons) by observation of the rate of reaction as a function of buffer concentration. See also catalysis, catalytic coefficient, intramolecular catalysis, pseudo-catalysis, specific catalysis.
kHA[HA] multiplied by some function of substrate concentrations. (The acids HA are unchanged by the overall reaction.) General catalysis by acids can be experimentally distinguished from specific catalysis by hydrogen cations (hydrons) by observation of the rate of reaction as a function of buffer concentration. See also catalysis, catalytic coefficient, intramolecular catalysis, pseudo-catalysis, specific catalysis.
The catalysis of a chemical reaction by a series of Brønsted bases (which may include the lyate ion) so that the rate of the catalysed part of the reaction is given by kB[B] multiplied by some function of substrate concentration. See also general acid catalysis.
A diagram showing the relative standard Gibbs energies of reactants, transition states, reaction intermediates, and products, in the same sequence as they occur in a chemical reaction. These points are often connected by a smooth curve (a "Gibbs energy profile", commonly still referred to as a "free energy profile") but experimental observation can provide information on relative standard Gibbs energies only at the maxima and minima and not at the configurations between them. The abscissa expresses the sequence of reactants, products, reaction intermediates and transition states and is usually undefined or only vaguely defined by the reaction coordinate (extent of bond breaking or bond making). In some adaptations the abscissas are however explicitly defined as bond orders, Brønsted exponents, etc.
Contrary to statements in many text books, the highest point on a Gibbs energy diagram does not necessarily correspond to the transition state of the rate-limiting step. For example, in a stepwise reaction consisting of two reaction steps
(1) A + BC
(2) C + D
E
one of the transition states of the two reaction steps must (in general) have a higher standard Gibbs energy than the other, whatever the concentration of D in the system. However, the value of that concentration will determine which of the reaction steps is rate-limiting. If the particular concentrations of interest, which may vary, are chosen as the standard state, then the rate-limiting step is the one of highest Gibbs energy.
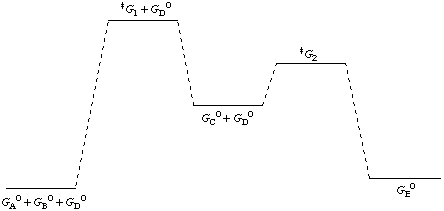
See also potential energy profile, potential energy (reaction) surface, reaction coordinate.
Gibbs energy of activation (standard free energy of activation), ![[double dagger]](../greek/DDAGGER.GIF) G (SI unit: kJ mol-1)
G (SI unit: kJ mol-1)
The standard Gibbs energy difference between the transition state of a reaction (either an elementary reaction or a stepwise reaction) and the ground state of the reactants. It is calculated from the experimental rate constant k via the conventional form of the absolute rate equation:
G = RT [ln(kB/h) - ln(k/T)]where kB is the Boltzmann constant and h the Planck constant (kB/h = 2.08358 x 1010 K-1 s-1). The values of the rate constants, and hence Gibbs energies of activation, depend upon the choice of concentration units (or of the thermodynamic standard state). See also enthalpy of activation, entropy of activation.
The state of lowest Gibbs energy of a system. IUPAC ANALYTICAL CHEMISTRY (1982). See also excited state.
A defined linked collection of atoms or a single atom within a molecular entity. This use of the term in physical organic and general chemistry is less restrictive than the definition adopted for the purpose of nomenclature of organic compounds. [See IUPAC ORGANIC RULES (1979), Section C]. See also substituent.
The linear free energy relation
expressing the dependence of the rate of solvolysis of a substrate on ionizing power of the solvent. The rate constant k0 applies to the reference solvent (ethanol-water, 80:20, v/v) and ks to the solvent s, both at 25 oC. The parameter m is characteristic of the substrate and is assigned the value unity for tert-butyl chloride. The value Y is intended to be a quantitative measure of the ionizing power of the solvent s. The equation was later extended by WINSTEIN, GRUNWALD and JONES (1951) to the form
where N is the nucleophilicity of the solvent and l its susceptibility parameter. The equation has also been applied to reactions other than solvolysis. [For the definition of other Y-scales, see BENTLEY and SCHLEYER (1977), BENTLEY and LLEWELLYN (1990).] GRUNWALD and WINSTEIN (1948); FAINBERG and WINSTEIN (1956). See also Dimroth-Reichardt ET parameter, polarity, Z-value.
An organic or inorganic ion or molecule that occupies a cavity, cleft or pocket within the molecular structure of a host molecular entity and forms a complex with it or that is trapped in a cavity within the crystal structure of a host. See also crown, cryptand, inclusion compound.
BENTLEY, T. W., and LLEWELLYN, G. (1990), Progr. Phys. Org. Chem., 17, 121-158.
BENTLEY, T. W., and SCHLEYER, P. v. R. (1977), Adv. Phys. Org. Chem., 14, 1-67.
FAINBERG A. H., and WINSTEIN, S. (1956), J. Am. Chem. Soc., 78, 2770-2777.
FLEMING, I. (1976), "Frontier Orbitals and Organic Chemical Reactions", Wiley, New York.
FUKUI, K., YONEZAWA, T., and SHINGU, H. (1952), J. Chem. Phys., 20, 722-725.
GOLD, V. (1969), Adv. Phys. Org. Chem., 7, 259-331.
GROB, C. A. (1969), Angew. Chem., Int. Ed. Engl., 8, 535-546.
GRUNWALD, E., and WINSTEIN, S. (1948), J. Am. Chem. Soc., 70, 846-854.
KIRKWOOD, J. G., and WESTHEIMER, F. H. (1938), J. Chem. Phys., 6, 506-512, 513-517.
WINSTEIN, S., GRUNWALD, E., and JONES, H. W. (1951), J. Am. Chem. Soc., 73, 2700-2707.
Return to home page for Glossary of terms used in Physical Organic Chemistry.